Single-Cell RNA-Seq Analysis Overview
For projects sequenced at our core facility, we offer in-depth single-cell RNA sequencing analysis specifically tailored to the project design. Because of the complex nature of this analysis, we advise sitting down with our team of bioinformaticians and planning out the analysis, prior to sequencing.
- 10X Genomics Cellranger pipeline
- Quality-Review
- Transcriptome Alignments
- R-based Seurat single-cell analysis
- Velocyto trajectory analysis
Additional analysis are available on a per-project basis, and subsequent follow up meetings between the researcher and data analyst is recommended and encouraged.
Linked-Read Whole Genome Analysis Overview
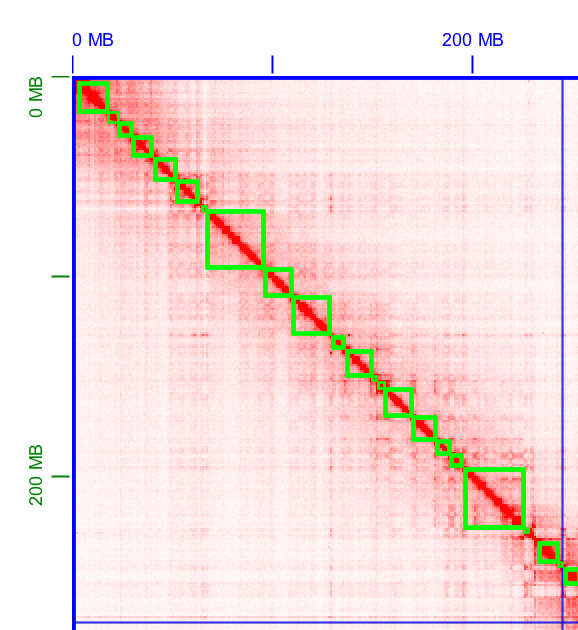 10X Genomics sample prep provides long-range information through short-read sequencing by introducing molecule-specific barcoding. This enables detection of phased-SNP, Indel, and structural variation in model organisms, as well as high-quality draft genome for denovo assembly projects. For denovo based assemblies, please contact us regarding the data analysis as this can be computationally intensive and the starting DNA material is key to downstream analysis success.
10X Genomics sample prep provides long-range information through short-read sequencing by introducing molecule-specific barcoding. This enables detection of phased-SNP, Indel, and structural variation in model organisms, as well as high-quality draft genome for denovo assembly projects. For denovo based assemblies, please contact us regarding the data analysis as this can be computationally intensive and the starting DNA material is key to downstream analysis success.
- 10X Genomics Longranger Pipeline
- 10X Genomics Supernova Pipeline
- Data integration with HiC libraries using Juicebox and 3d-dna assembly
ChIP-Seq Analysis Overview
We offer an affordable second tier analysis pipeline for your ChIP-Seq sequencing data. This analysis workflow includes the following protocols: 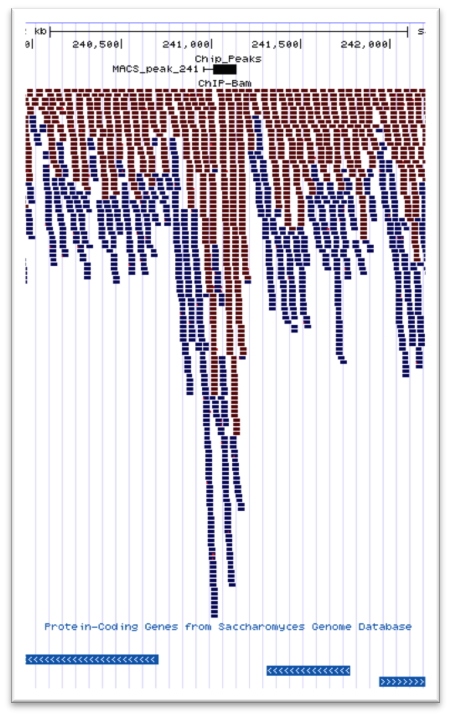
- FASTQC quality control.
- Alignment to a standard reference genome.
- NSC/RSC Summary Cross-correlation analysis
- Predicted binding sites using MACS2
- UCSC Visualization
With this output, you will be able to quickly dive into the biology behind your experiment. All of the analysis results will be accessible for download via a secure FTP server. A download link will be provided upon completion of the analysis workflow.
RNA-Seq Analysis Overview
Quantifying gene expression from RNA-Seq data requires several steps that are very time and resource intensive. We have worked to establish a best-practices pipeline for our core customers. This workflow produces:
- FASTQC quality control
- Tophat alignment to a standard reference genome
- Cufflinks Gene and Isoform level FPKM expression measurements
- Cuffdiff Case-Control differential expression testing
- UCSC Visualization
All of the analysis results will be accessible for download via a secure FTP server. A download link will be provided upon completion of the analysis workflow. If grouped comparisons are requested, a detailed explanation of the project’s experimental design is required to be submitted upon initial sample submission.
miRNA Analysis Package
Due to the short sequence length of miRNA(22 – 27nt), custom  trimming of the illumina adapter sequence is required before genome alignment and miRNA quantification. We use the commercial software package CLC Genomics Workbench to handle our miRNA analysis pipeline. This analysis includes:
trimming of the illumina adapter sequence is required before genome alignment and miRNA quantification. We use the commercial software package CLC Genomics Workbench to handle our miRNA analysis pipeline. This analysis includes:
- Custom Adapter Trimming from the 3′ end.
- Quantification of miRNA populations using mirBASE 20.
- Excel tables with mature 5′ and 3′ counts and exact mature
A download link will be provided upon completion of the analysis workflow. If grouped comparisons are requested, a detailed explanation of the project’s experimental design is required to be submitted upon initial sample submission.
Custom Analysis
Many biological questions persist after initial data analysis. For specific requests, an in-person meeting at the Center for Bioinformatics and Life Sciences with the UB GBC staff members is encouraged. Typically we require at least a two- week lead-time for in-person meetings, with experimental information provided prior to meeting with us.
Please note, we also take requests for analysis of data generated outside of our facility. If you have data and are having difficulty, please feel free to contact us with any questions!
Á La Carte
Alignments
Data sequenced at our facility can be taken from raw fastq to aligned BAM files via Bowtie/Bowtie2/BWA.
Peak Calling
ChIP-Seq samples can be processed for peak calling through the MACS2 algorithm.
Bacterial Variant Calling
CLC Genomics variant calling for bacterial samples. Includes SNP and Small Indel calling.
Authorship
In accordance with ABRF’S Authorship Guidelines, we request co-authorship if we have developed tools, algorithms, or pipelines, participated in experiment design, or contributed to a biological question addressed in a manuscript.
For all projects sequenced and analyzed by our core facility, we request, at minimum, you acknowledge us in a publication to which we have contributed routine analysis, or data management and storage.